Molecular weight analysis¶
RAW provides four forms of molecular weight analysis:
- Referencing I(0) to that of a known standard
- From the volume of correlation using the method of Rambo and Tainer
- From the adjusted Porod volume using the method of Fisher et al.
- From the value of I(0) on an absolute scale.
In RAW, right click on the subtracted GI scattering profile in the manipulation panel and select “Molecular weight.” At the top of the panel is a description of the methods used, and the results of your Guinier fit. All four methods require a good Guinier fit, so you can use that button to redo the fit if necessary. In the lower part of the panel, the results of the four estimates for MW are shown.
- Note: Neither the I(0) Ref. MW panel nor the Abs. MW panel should be reporting a MW.
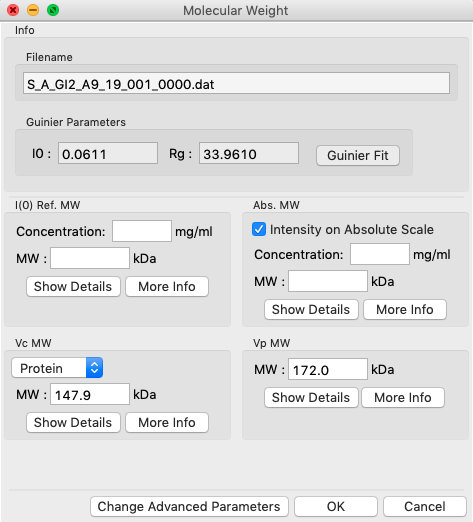
In either concentration box, enter the sample concentration of 0.47 mg/ml. Notice that you now get results from all four methods of MW calculation.
- Question: The expected MW value for GI is 172 kDa. How do your results compare?
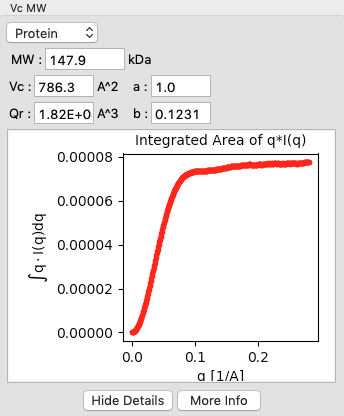
Click on the “Show Details” button for the Vc MW panel. You should see a graph, which shows the integrated area of \(qI(q)\) vs. q. For this method to be accurate, this value needs to converge at high q.
Click the “OK” button to save your analysis.
- Note: The “Cancel” button discards the analysis.
Repeat the MW analysis for the lysozyme sample, which had a concentration of 4.27 mg/ml. The expected MW of lysozyme is 14.3 kDa.
- Question: Does the Vc method work for the lysozyme data?
Aside: Discussion of the four methods:
Calculation of MW from reference to a known protein standard
The scattering at zero angle, I(0) is proportional to the molecular weight of the macromolecule, and the concentration and contrast of the macromolecule in solution. If a reference sample of known molecular weight and concentration is measured, it can be used to calibrate the molecular weight of any other scattering profile with known concentration (assuming constant contrast between reference and sample, and a monodisperse sample).
This method can yield inaccurate results if:
- The reference is not properly calibrated (concentration, I(0) measurement).
- I(0) is poorly determined.
- Sample concentration is poorly determined.
- The contrast between the macromolecule and buffer is significantly different between the reference and sample.
Calculation of MW from absolute scale This uses the absolute calibration of the scattering profile to determine the molecular weight, as described in Orthaber, D., Bergmann, A., & Glatter, O. (2000). J. Appl. Crystallogr. 33, 218-225.
This method can yield inaccurate results if:
- The absolute calibration is not accurate.
- I(0) is poorly determined.
- Sample concentration is poorly determined.
- Scattering contrast per unit mass is wrong. This depends on the buffer, macromolecule type (protein vs. nucleic acid), and the macromolecule partial specific volume (which can depend on shape/flexibility). The defaults are for a buffer with the electron density of water and compact globular proteins.
Volume of Correlation method
This method uses the approach described in: Rambo, R. P. & Tainer, J. A. (2013). Nature. 496, 477-481. This method should work for both compact and flexible macromolecules. The authors claim the error in MW determination is ~5-10%.
This method can yield inaccurate results if:
- The integral of q*I(q) doesn’t converge (click ‘Show Details’ to see), which can indicate the scattering profile is not measured to high enough q or that there is a bad buffer match.
- I(0) and/or Rg are poorly determined.
- You have a protein-nucleic acid complex.
- Your molecule is less than ~15-20 kDa.
Adjusted Porod Volume method
This method uses the approach described in: V. Piiadov, E. Ares de Araujo, M. Oliveira Neto, A. F. Craievich, and I. Polikarpov. Protein Science (2019). 28(2), 454-473. It applies a correction to the Porod volume for the finite length of the measurement. The authors report a median of 12% uncertainty for calculated molecular weight from globular proteins.
This method can yield inaccurate results if:
- The molecule is not globular (i.e. is flexible or extended).
- I(0) is poorly determined.
- The protein density used is inaccurate (can be changed).
- Your molecule is not a protein (e.g. RNA/DNA or a protein-nucleic acid complex).