Pair-distance distribution analysis – GNOM in RAW¶
The first step in most advanced data processing is to calculate the P(r) function, the inverse Fourier transform of I(q). This cannot be calculated directly from the scattering profile, so indirect Fourier transform methods are typically used. The most common such method is implemented in the GNOM program from the ATSAS package. We will use RAW to run GNOM. Note that you need ATSAS installed to do this part of the tutorial.
Open RAW. The install instructions contain information on installing and running RAW. If RAW is already open, clear all loaded data in RAW.
Open the lysozyme.dat file in the Tutorial_Data/reconstruction_data folder.
Right click on the lysozyme profile in the Manipulation list and select “IFT (GNOM)”.
- Note: RAW will automatically try to find an appropriate maximum dimension (Dmax) by running the DATGNOM program from the ATSAS software package.
- Troubleshooting: If you do not have the GNOM option in the right click menu, RAW does not know where your ATSAS programs are installed. If you installed the ATSAS programs after starting RAW, restart RAW and it will attempt to automatically find them. If that has failed, go to the Options->Advanced Options menu and choose the ATSAS settings (“ATSAS”). Uncheck the option to “Automatically find the ATSAS bin location”, and specify the location manually.
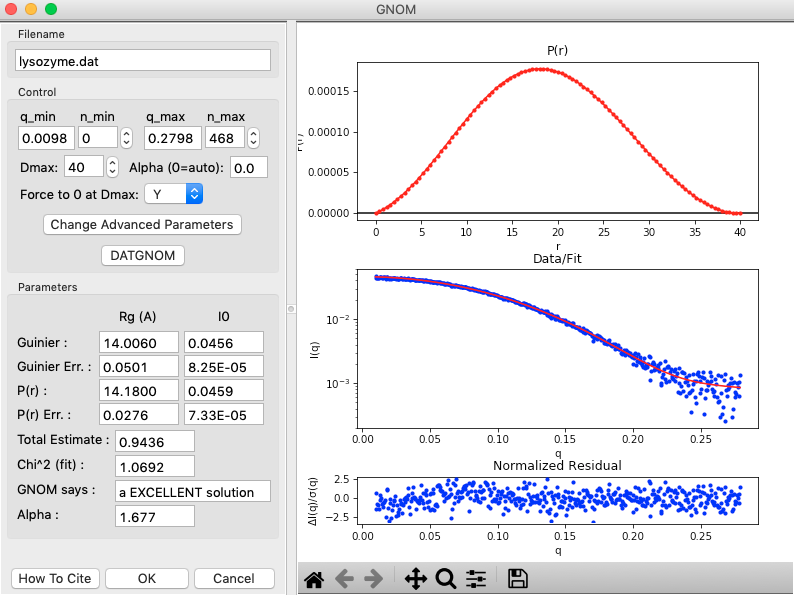
The GNOM panel has plots on the right. These show the P(r) function (top panel), the data (middle panel, blue points) and the fit line (middle panel, red line), and the fit residual (bottom panel).
- Note: The fit line is the Fourier transform of the P(r) function, and is also called the regularized intensity.
On the left of the GNOM panel are the controls and the resulting parameters. You can alter the data range used, the Dmax value, and the alpha value used.
- Tip: The Guinier and P(r) Rg and I(0) values should agree well. The total estimate varies from 0 to 1, with 1 being ideal. GNOM also provides an estimate of the quality of the solution. You want it to be at least a “REASONABLE” solution.
Vary the Dmax value up and down in the range of 30-50 in steps of 1. Observe what happens to the P(r) and the quality of the solution.
- Note: Dmax is in units of Å.
- Tip: Recall that we want the following qualities in a P(R) function:
- No negative values.
- A gentle approach to Dmax (not suddenly forced down).
- Minimal oscillation.
Return the Dmax value to that found by DATGNOM by clicking the “DATGNOM” button. Dmax should be 40. By default, GNOM forces the P(r) function to zero at Dmax. For a high quality data set and a good choice of Dmax, P(r) should go to zero naturally. Change the “Force to 0 at Dmax” option to “N”.
- Try: Vary Dmax with this option turned off.
Reset it so that the P(r) function is again being forced to zero at Dmax.
Set the Dmax back to 40, and click OK. This saves the results into the RAW IFT panel.
Click on the IFT Control and Plot tabs. This will display the GNOM output you just generated. Save the lysozyme.out item in the reconstruction_data folder.
- Note: This saved file is all of the GNOM output, in the GNOM format. It can be used as input for any program that needs a GNOM .out file.