RAW Settings and the Options window¶
WARNING: The manual is current several versions out of date. While it may still be useful for some users, please refer to the tutorial for the most up-to-date information.
This section covers the RAW settings, in particular focusing on the Options window and what all of the settings there do. It also covers how to save and load settings in RAW.
Saving and loading settings¶
To save the current RAW settings to disk (a .cfg file):
- Go to the “File” menu bar and select “Save Settings”
- Select a filename and a location for the configuration (config) file and click “Save”
To load RAW settings from disk (a .cfg file):
- Go to the “File” menu bar and select “Load Settings”
- Select the appropriate file and click “Open”
Note: Saving a configuration file saves all of the settings in RAW that are set in the Options window. It also saves any masks that have been created.
Changing settings¶
Settings in RAW are generally changed in the Options window. Below we describe how to open the options window, how to change all of the settings, and what the settings do.
Opening the Options window¶
To open the options window:
- Go to the “Options” menu
- Select “Advanced Options”
There are two parts to the Options window. There is an options tree on the left that determines which options panel is displayed on the right. The part on the right displays the options associated with the selected option in the option tree.
Closing the Options window¶
To save the changes made to the settings in the Options window, close the window by clicking the “OK” button. To exit without saving any of the changes in the Options window, use the “Cancel” button or the system close button (an “x” in the upper left or right corner of the panel).
General settings panel¶
The general settings are all check boxes and can be set by checking and unchecking the boxes. They control the following items:
Hide controls on manipulation items for new plots
If this option is selected, new Manipulation data items start out collapsed, if not selected they start out expanded. Defaults to off.
Write header on top of dat files
The .dat and .ift file “header” information can be saved at the top or bottom of the file. If this is selected, it is written at the top, if not it is written at the bottom. Defaults to off.
Use header for mask creation (SAXSLAB instruments)
If this option is set, if a SAXSLab instrument is being used, the image header will be used to make the beamstop mask. Defaults to off.
Detector is rotated 90 degrees (SAXSLAB instruments)
If this option is set, it indicates to RAW that the SAXSLAB detector is rotated 90 degrees. This affects mask creation from the image header. Defaults to off.
Start online mode on startup
If this option is selected and an Online mode startup directory has been picked, then when RAW starts, it will automatically turn on online mode, with the selected directory as the target online directory.
2D Reduction¶
In the 2D reduction panel, there are reduction options.
Correct of the change in the solid angle of the pixels
If this option is selected, the scattering intensity is corrected for the change in solid angle of pixels as you move along the detector. This is implemented as the standard \(\cos^3 (2\theta)\) where \(2\theta\) is the scattering angle.
Image/Header Format panel¶
This panel in the Options window allows you to set the image format, the header file format (if any), load the image header into RAW to set up normalizations, and set up bindings to use information in the image header or header file as calibration and reduction parameters.
Image format¶
The image format can be set using the drop down menu. A full description of supported image types is available in the file types section.
Header file format¶
The header file is a file that accompanies the detector image on some beamlines. This file often contains additional information such as, diode values, ion chamber readings, exposure time and date. Currently RAW supports the following header files: I711, MAXLab; I911-4, MAXLab; F2, CHESS; G1, CHESS; G1 WAXS, CHESS; G1 Eiger, CHESS; BL19U2, SSRF; and BioCAT, APS.
Note: If you wish to have a new header file format added to RAW, please contact the developers.
Loading/Viewing header information¶
If you wish to view header information from either the header file or the image header, click the “Load Image” button and select the image of interest.
If you wish to use the header information to normalize the image, load the image using the “Load Image” button and then click the “Apply” button at the bottom of the screen. This will save the counter values in such a way that RAW can set up the normalization appropriately.
Using image header information for calibration and reduction (turning on and setting bindings)¶
RAW has the ability to use header information for calibration and reduction settings. The method for doing this is to set a “binding” between the counter value and the calibration value. The calibration values that can be obtained from the image header or header file are: Beam X Center, Beam Y Center, Detector Pixel Size, Sample Detector Distance, and Wavelength.
To create a binding:
- Check the “Use image-header/header file for calibration and reduction parameters” box.
- Load the image and header file values into the list as described above.
- In the list of the image header and header file names and values, click on the name of the header parameter that you want to use as one of the calibration values. This will fill in the Name and Value in the appropriate fields in the lower left hand portion of the panel.
- Using the “Binding” menu, select what calibration parameter should use this header value. In the binding column of the header list, you will see this calibration parameter displayed.
Note: These values overwrite the same values set elsewhere in the settings. So if you bind the Beam X Center to use a value from the header, no matter what you set it to in the Calibration panel of the Options window it will use the value from the header.
Note 2: Make sure that your header file values match the expected units for the calibration parameter. The beam center values should be in pixels on the detector, the detector pixel size should be in microns, the sample detector distance in mm, and the wavelength in angstroms.
Adding a modifier to a binding¶
Once a binding is set, it is possible to add a modifier to the binding, which affects the value obtained from the header. This might be used in a case where the header value is not in the appropriate units.
To set a modifier:
- In the list of the image header and header file names and values, click on the name of the bound header parameter that you want to set a modifier for.
- In the Modifier field at the bottom of the panel, type in a mathematical expression. This expression may contain any of the header values (including but not limited to the header value selected for the binding). It may contain “+” “-“ “*” and “/” for addition, subtraction, multiplication, and division. The following strings are restricted, and apply specific mathematical functions: acos, asin, atan, atan2, ceil, cos, cosh, degrees, exp, fabs, floor, fmod, frexp, hypot, ldexp, log, log10, modf, pow, radians, sin, sinh, sqrt, tan, tanh, all of which correspond to the functions of the same name in the python math library ( https://docs.python.org/2/library/math.html#module-math ).
- Click the “Add” button. You should get a popup window that evaluates the expression for the current loaded header values. Once you close that window, the modifier should be listed in the Modifer column of the header list.
Changing or removing a modifier to a binding¶
To change a modifier to a binding, do the steps to add a modifier, above. When you click on the header item in step 1, the modifier will be shown in the Modifier field at the bottom of the panel, and you can make changes as appropriate in step 2.
To remove a modifier to a binding:
- In the list of the image header and header file names and values, click on the name of the bound header parameter that you want to remove a modifier to.
- Click the “Remove” button (next to the Modifier field at the bottom of the panel).
Removing bindings¶
To remove a single binding:
- In the list of the image header and header file names and values, click on the name of the bound header parameter that you want to remove a binding to.
- In the “Binding” menu at the bottom of the panel, select “No binding”.
To remove all bindings, click the “Clear Bindings” button.
Disabling bindings for calibration and reduction¶
To disable the use of bindings for calibration and reduction, either remove all bindings or uncheck the “Use image-header/header file for calibration and reduction parameters” checkbox.
Clear All¶
The “Clear All” button clears all bindings, and removes the current loaded header/header file values from the panel.
Calibration panel¶
The calibration panel allows you to set the beam center, binning size, number of points skipped at the start and end of a scattering profile, the sample to detector distance, wavelength, detector pixel size, and whether or not the Q range is calibrated.
Setting calibration parameters¶
The calibration paramters are: Beam center (x and y), sample-detector distance, wavelength, and detector pixel size. These can all be set by entering a value in the appropriate field on this panel or using the spin controls. However, it is more natural to set these values from the Calibration/Centering panel.
Note: Changing the settings in the calibration/centering panel will change the values in this panel, and vice versa.
Note 2: All of these calibration values are overridden by the bindings described above, if a binding for the particular calibration parameter is set.
Start and end points¶
The “Start plots at q point number” value sets the first q point shown on the plot when a scattering profile is integrated. It is zero indexed (first point is zero). So if it is set to 5, the plot will start with the 6th q point in the q-vector. This is typically used to get rid of the beamstop shadow from the integrated profiles.
The “Skip n points at the end of the curve” value sets the last point shown on the plot when a scattering profile is integrated. If it is set to zero, all points are shown. So if it is set to 5, the last point shown will be the 5th to last point in the q-vector. This is typically used to remove end points if something, for example the downstream flight tube window, is shadowing a high q region of the detector.
Binning¶
The default binning for integrated scattering profiles can be set using the “Binning Size” option. It accepts integer values. A binning size of one corresponds to q bins that are one pixel wide. A binning size of 2 corresponds to q bins that are 2 pixels wide, and so on.
Note: The q size of a bin of a given pixel size will depend on the calibration parameters.
Calibrating the q-range¶
If you do not wish to calibrate the q-range of integrated scattering profiles, uncheck the “Calibrate Q-range” box. The scattering profile will then be displayed as intensity vs. bin number. This option is checked by default.
Normalization panel¶
The normalization panel allows you to normalize integrated scattering profiles by some value. Typically a counter value is used that is proportional to the beam intensity transmitted through the sample (such as a beamstop counter from an active beamstop).
Enabling and disabling normalization¶
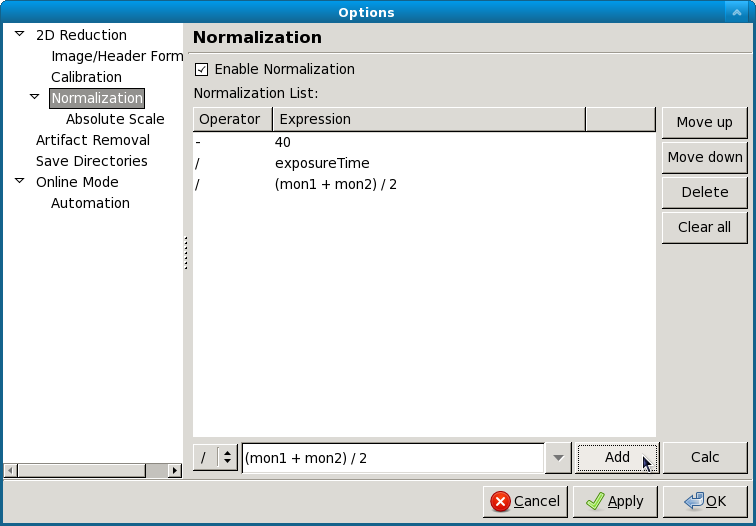
To enable normalization for integrated scattering profiles, check the “Enable Normalization” checkbox (checked by default). To disable, uncheck the “Enable Normalization” checkbox.
Setting up normalization operations¶
To add a new operation to the normalization procedure:
- Select the operator to be used (/, *, -, + corresponding to division, multiplication, subtraction, and addition respectively)
- Enter the desired expression in the expression box.
- Click the “Calc” button to view the result of the entered expression.
- Click the “Add” button.
Note: This expression may contain any of the header values (including but not limited to the header value selected for the binding). It may contain “+” “-“ “*” and “/” for addition, subtraction, multiplication, and division. The following strings are restricted, and apply specific mathematical functions: acos, asin, atan, atan2, ceil, cos, cosh, degrees, exp, fabs, floor, fmod, frexp, hypot, ldexp, log, log10, modf, pow, radians, sin, sinh, sqrt, tan, tanh, all of which correspond to the functions of the same name in the python math library ( https://docs.python.org/2/library/math.html#module-math ).
Reordering and removing normalization operations¶
The order in which the operations are carried out can be changed by selecting the operation in the normalization list and using the Move Up and Move Down buttons. Operations can be removed by selecting the operation in the list and clicking the Delete button.
Normalizing by a header value¶
It is often desired to normalize the data by exposure time or incoming / transmitted beam intensity, and/or remove offsets on the detector.
To do so:
- Load a header file into RAW as described. Be sure to hit the “Apply” button after loading!
- Return to the Normalization panel.
- Add a normalization value as in steps, in the expression box enter the name of the header value you wish to normalize by along with any other mathematical operations.
Normalizing by a region of interest (ROI)¶
RAW has the ability to normalize by a region of interest on the image. Every pixel in the region of interest is summed, and that can be used to normalize in the same way as a header value.
To normalize by an ROI:
- Set an ROI mask.
- Add an operation to the normalization list, but use “roi_counter” (without quotes) as the header value. For example, to divide by the roi value, select the “/” operator and enter roi_counter in the expression box, then add that to the list.
Absolute scale panel¶
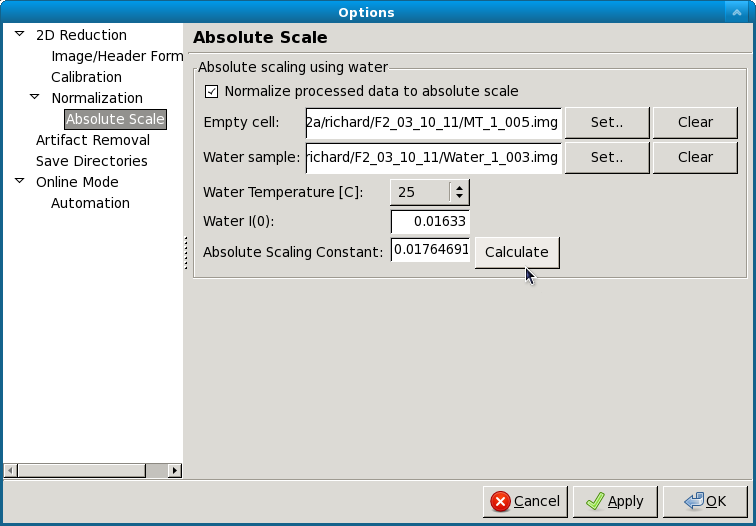
RAW is able to scale loaded image data to absolute scale using water as a standard. Water has a known, temperature dependent absolute scale value at the forward scattering I(0). Water has a relatively flat scattering profile, which makes it possible to estimate the forward scattering, I(0), from an average of the intensity. To obtain the pure water signal, the water sample obtained in a sample cell must have the empty cell subtracted from it.
To set up Absolute scale:
- Click the “Set” button for the empty cell. Select either an image or text (such as .dat) file of the empty cell scattering.
- Click the “Set” button for the water sample. Select either an image or text (such as .dat) file of the water scattering.
- Select the water temperature in degrees centigrade.
- Click the “Calculate” button. An absolute scaling constant should appear.
- Enable absolute scale normalization by checking “Normalize processed data to absolute scale” check box.
The algorithm uses the middle third part of the water scattering curve to estimate I(0) by the average intensity.
Note: The selected files must have been normalized in exactly the same way as the rest of the data that is to go on absolute scale. If loading an image, that means not changing the normalization parameters after calculating the absolute scale. If normalization parameters are changed the absolute scale constant will have to be re-calculated. It is particularly important that the images or profiles used to calculate absolute scale not have been saved with absolute scale already on (for example, from a previous calibration).
Turning off absolute scale¶
To turn off absolute scale, uncheck the “Normalize processed data to absolute scale” checkbox in the Absolute scale panel.
Flatfield correction panel¶
If a flatfield file is available, RAW can do a flatfield correction of the data. To do so, click the “Set” button, and select the flatfield image. Then check the “Enable flatfield correction” box.
When RAW applies a flatfield correction, it divides every image it processes by the flatfield image, on a per-pixel basis. The assumption is that every pixel in the flatfield image should have gotten the same intensity, so any variation comes from variation in the detector.
Turning off flatfield correction¶
To turn off flatfield correction, uncheck the “Enable flatfield correction” checkbox in the Flatfield correction panel.
Molecular weight panel¶
The molecular weight panel of the Options windows allows control of the parameters used to calculate molecular weight in the molecular weight window and the SEC calculated parameters. All four methods are described in more detail elsewhere.
Molecular Weight Estimation Using a Standard
This subpanel corresponds to parameters for the MW estimation by comparison to a known standard. While all of the parameters of the standard can be set/changed in this box, the standard MW (in kDa), the standard I(0), the standard concentration (in mg/ml), and the standard filename (only for reference), it is more natural to change these settings by loading the standard scattering profile into RAW and using the Use as MW Standard option.
Molecular Weight Estimation From Volume of Correlation
This subpanel corresponds to the parameters used for the volume of correlation method of estimating molecular weight. This method is the method used for calculating MW in the SEC panel ). The protein and RNA coefficients correspond to the A and B coefficients. The default type selection selects if the MW calculation defaults to Protein or RNA. Changing this option will change whether the MW calculated in the SEC panel is for protein or RNA.
Molecular Weight Estimation From Corrected Porod Volume
This subpanel corresponds to the parameters for the MW calculation by corrected Porod volume. For this method, the only parmater that can be changed is the protein density in kDa/Å:sup:3.
Molecular Weight Estimation From Absolute Intensity Calibration
These parameters correspond to the parameters necessary for calculating the molecular weight when a scattering profile is on an absolute scale.
Reset MW Parameters To Defaults
If you have customized the MW parameters for a particular sample, you can restore the parameters to the RAW defaults (which are the defaults from the relevant papers for each method). There are no default settings for the estimation using a standard.
Artifact removal panel¶
Zingers are pixel values on the detector that are unusually high due to either cosmic radiation or readout errors. RAW includes three methods that can be used for zinger removal.
Zinger removal by smoothing¶
A window of “Window Length” data points can be run across the data and discard values that are more than “Std” standard deviations away from the average of the points in the window. A starting index is given to specify where on the data curve the window should start.
Zinger removal when averaging¶
If three or more exposures of the same sample are available, then these can be used to eliminate zingers by comparing the intensity values of each data set to the others. An intensity value in a data-set that is larger than x standard deviations (Sensitivity) from third quintile of all related data points in the rest of the data sets is removed and replaced by the average of the third quintile.
Zinger removal after radial averaging¶
This method is the most effective method for removing zingers. Pixel intensities in the image for the same q value are compared and should be fairly constant. Values that are more than “Sensitivity” standard deviations away from the median are discarded.
IFT panel¶
RAW currently supports one built-in method for determining the inverse Fourier transform (IFT) of a scattering profile, the Bayesian IFT (BIFT) method. In the future we anticipate supporting a python based implementation of the GNOM algorithm called pyGNOM, but currently that is not available.
BIFT¶
The BIFT panel allows you to set the BIFT Grid-Search parameters. These define the large grid that the BIFT algorithm searches over before doing the fine search near the best value on the grid.
Dmax Upper Bound
Sets the largest maximum dimension (Dmax) value that will be used in the coarse grid search, in Å.
Dmax Lower Bound
Sets the smallest Dmax value that will be used in the coarse grid search, in Å.
Dmax Search Points
Total number of Dmax values in the coarse grid search that. These are evenly distributed between the lower and upper bounds.
Alpha Upper Bound
Sets the largest alpha value that will be used in the coarse grid search.
Alpha Lower Bound
Sets the smallest alpha value that will be used in the coarse grid search.
Alpha search points
Sets the total number of alpha values in the coarse grid search. These are distributed logarithmically between the lower and upper bound.
P(r) Points
Sets the number of points in the calculated P(r) curve.
Save Directories panel¶
This panel controls the settings for automated saving of data.
Auto Save
In this subpanel, the checkboxes control whether or not RAW automatically saves Processed image files, Averaged data files, and Subtracted data files. When the boxes are checked, that file type will be automatically saved, when they are unchecked, it will not.
Save Directories
This panel allows you to selected the directories into which files will be saved for each of the automated saving file types (Processed, Averaged, Subtracted). To pick a directory, click the “Set” button and use the window that opens to select a folder. Click “Open” once the appropriate folder is selected. To clear a directory click the “ Clear” button.
Note: A save directory must be selected before an Auto Save checkbox can be enabled.
Online Mode panel¶
This panel includes settings for the online mode. This lets you enable online filtering and set up the filter list.
Enabling/disabling online filtering¶
Online filtering filters files by filename, so that you can control which files are loaded into RAW automatically. To enable this mode, check the “Enable Online Filtering” checkbox. To disable this mode, uncheck that checkbox.
Adding a filter item to the online filter list¶
A filter item consists of three parts. First, there is the Ignore/Open operator. This allows you specify whether you want RAW to ignore files with the given filter string in their name, or to only open files that have the given filter string in their name. To set this option, use the dropdown selector box at the bottom of the panel and select either “Ignore” or “Open only with”.
The next part of the filter is the filter string. This is the string that RAW looks for in the filename. To set this, enter a string into the filter string box at the bottom of the panel.
The final part of the filter is the location of the filter string. This sets where in the filename RAW looks for the given filter string. This can be set to: “At start”, which means RAW only applies the filter Ignore/Open action to files with the filter string at the start of the file name; “Anywhere”, which means RAW applies the filter Ignore/Open action to files with the filter string anywhere in the file name; and “At end”, which means RAW only applies the filter Ignore/Open action to files with the filter string at the end of the file name. To set this, use the dropdown selector box at the bottom of the panel and select one of those three options.
Once you have set the Ignore/Open option, entered a filter string, and selected the location of the filter string, click the “Add” button to add the filter item to the Online Filter List.
Reordering and removing filter items¶
The order in which the filtering is carried out can be changed by selecting the item in the filter list and using the Move Up and Move Down buttons. Items can be removed by selecting the operation in the list and clicking the Delete button. All filter items can be removed using the “Clear all” button.
SEC-SAXS panel¶
The SEC-SAXS panel controls settings related to the SEC-SAXS data processing.
Intensity ratio (to background) threshold for calculationg Rg, MW, I0
In order to speed up the calculation of Rg, MW, and I0 as a function of frame for SEC-SAXS data, a ratio of the frame intensity to the background intensity is taken. If that value is less than the threshold set here, the frame is skipped. To attempt to calculate structural parameters for all frames, set this threshold to -1.
ATSAS panel¶
The top level ATSAS panel allows you to control where the ATSAS bin location is (the folder with all of the ATSAS programs in it). By default, RAW will attempt to automatically find the ATSAS installation. If you wish to set the location yourself, uncheck the “Automatically find the ATSAS bin location” checkbox, and either type the location into the ATSAS bin location field or use the “Select Directory” button to select the appropriate directory.
Note: If you uncheck and then check the Automatically find checkbox, RAW will attempt to find the ATSAS directory again. This can be useful if, for example, you install ATSAS and want RAW to find the new installation without restarting RAW.
GNOM panel¶
The top level GNOM panel allows you to set the commonly used advanced settings for the ATSAS software GNOM, which is run from the GNOM window. All of these options correspond in name and allowable values to those of GNOM as described in the GNOM manual: https://www.embl-hamburg.de/biosaxs/manuals/gnom.html
Settings can be rest to their defaults (which correspond to the GNOM defaults) by clicking the “Reset to default” button. This resets the settings in this panel and in the GNOM Advanced panel.
GNOM Advanced panel¶
This GNOM panel allows setting GNOM settings which are not as commonly used in GNOM. Again, all of the options correspond in name and allowable values to those of GNOM as described in the GNOM manual: https://www.embl-hamburg.de/biosaxs/manuals/gnom.html
Settings can be rest to their defaults (which correspond to the GNOM defaults) by clicking the “Reset to default” button in the GNOM panel. This resets the settings in this panel and in the GNOM panel.
DAMMIF panel¶
The top level DAMMIF panel allows setting two things: First, the default settings for DAMMIF that are set when the DAMMIF window is opened. Second, standard settings that are available in Fast and Slow mode can be set in the “Standard Settings” subpanel. All of the settings correspond in name and allowable values to those in the DAMMIF manual: https://www.embl-hamburg.de/biosaxs/manuals/dammif.html
Settings can be rest to their defaults (which generally correspond to the DAMMIF defaults) by clicking the “Reset to default” button. This resets the settings in this panel and in the DAMMIF Advanced panel.
DAMMIF Advanced panel¶
The settings in the DAMMIF advanced panel are only used when the “Custom” mode is selected in the DAMMIF panel. This is equivalent to the interactive DAMMIF mode at the command line. Unless otherwise noted, a value of -1 for a field indicates that it will use the default setting. The settings correspond in name and allowable values to those in the DAMMIF manual: https://www.embl-hamburg.de/biosaxs/manuals/dammif.html
Settings can be rest to their defaults (which generally correspond to the DAMMIF defaults) by clicking the “Reset to default” button in the DAMMIF panel. This resets the settings in this panel and in the DAMMIF panel.