Molecular weight analysis
This tutorial covers how to use RAW for molecular weight analysis. This is not a tutorial on basic principles and best practices for molecular weight analysis. For that, please see the SAXS tutorial.
RAW provides four methods of molecular weight analysis:
Referencing I(0) to that of a known standard
From the volume of correlation using the method of Rambo and Tainer
From the adjusted Porod volume using the method of Fisher et al.
From the value of I(0) on an absolute scale.
If ATSAS is installed, RAW also provides an additional two methods of MW calculation using the ATSAS tools, for a total of six different methods:
From classification by machine learning (ATSAS datclass/Shape&Size)
From a Bayesian estimation based on concentration independent methods (ATSAS datmw bayes)
If you use these MW methods in RAW, in addition to the RAW paper please cite these papers:
Volume of correlation: Rambo, R.P. & Tainer, J.A. Nature (2013). 496, 477-481. DOI: 10.1038/nature12070
Corrected Porod volume: V. Piiadov, E. Ares de Araujo, M. Oliveira Neto, A. F. Craievich, and I. Polikarpov. Protein Science (2019). 28(2), 454-473. DOI: 10.1002/pro.3528
Bayesian inference (ATSAS): Hajizadeh, N. R., Franke, D., Jeffries, C. M. & Svergun, D. I. (2018). Sci. Rep. 8, 7204. DOI: 10.1038/s41598-018-25355-2
Shape&Size (ATSAS): Franke, D., Jeffries, C. M. & Svergun, D. I. (2018). Biophys. J. 114, 2485–2492. DOI: 10.1016/j.bpj.2018.04.018
A video version of this tutorial is available:
The written version of the tutorial follows.
In RAW, right click on the subtracted GI scattering profile in the Profiles panel and select “Molecular weight.” Alternatively click on the “Mol. Weight” button at the bottom of the Profiles panel.

At the top of the panel are the results of the Guinier fit. All methods require a good Guinier fit, so you can use that button to redo the fit if necessary. In the lower part of the panel, the results of the estimates for MW are shown.
Note: Neither the I(0) Ref. MW panel nor the Abs. MW panel should be reporting a MW.
Note: If you have ATSAS installed and accessible to RAW, you should see six panels with MW, as in the image below. If you don’t have ATSAS installed you will see just four panels, the rightmost panel in each row will be missing.
Tip: To learn more about any of the methods, click on the “More Info” button.
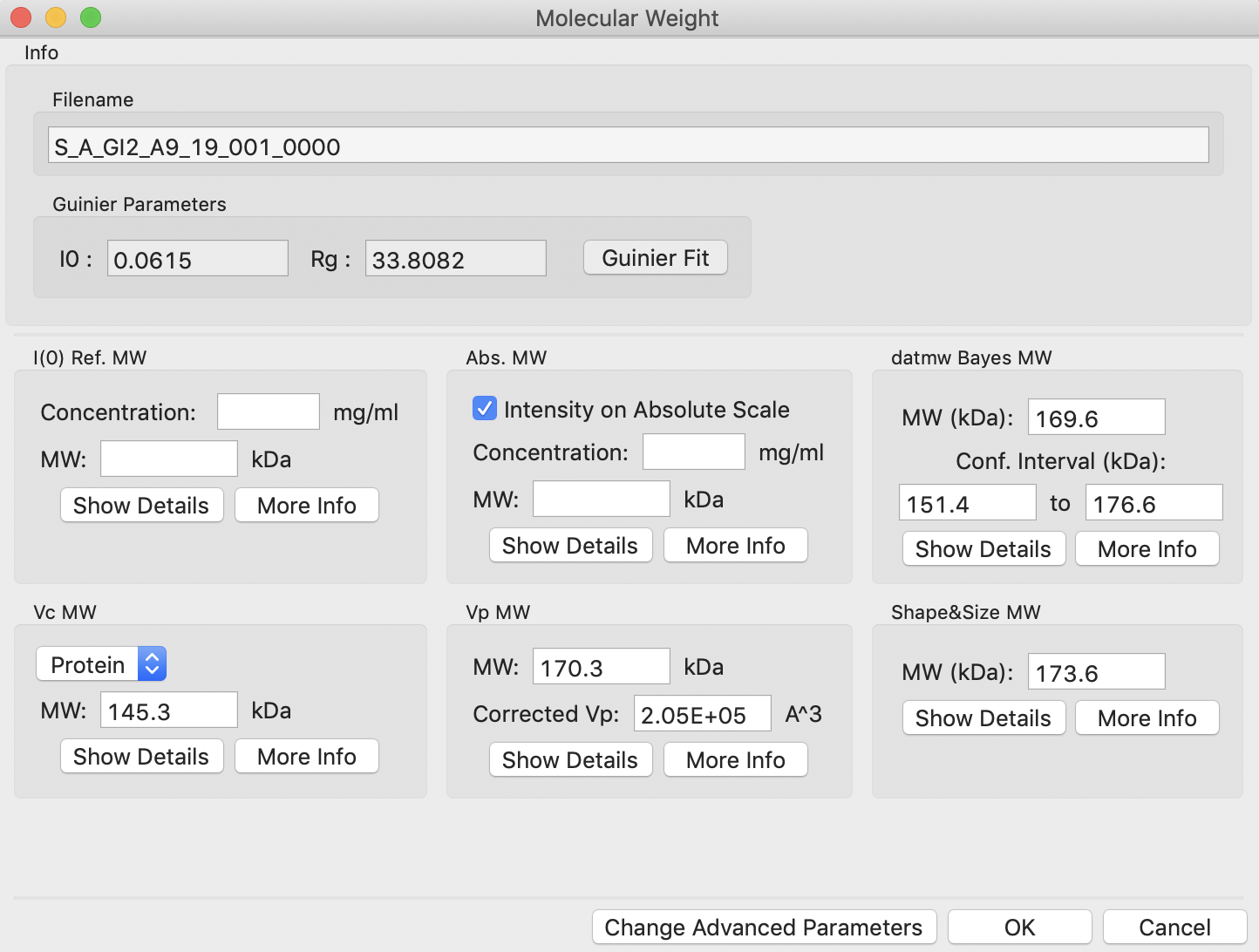
In either concentration box, enter the sample concentration of 0.47 mg/ml. Notice that you now get results from all methods of MW calculation.
Question: The expected MW value for GI is 172 kDa. How do your results compare?
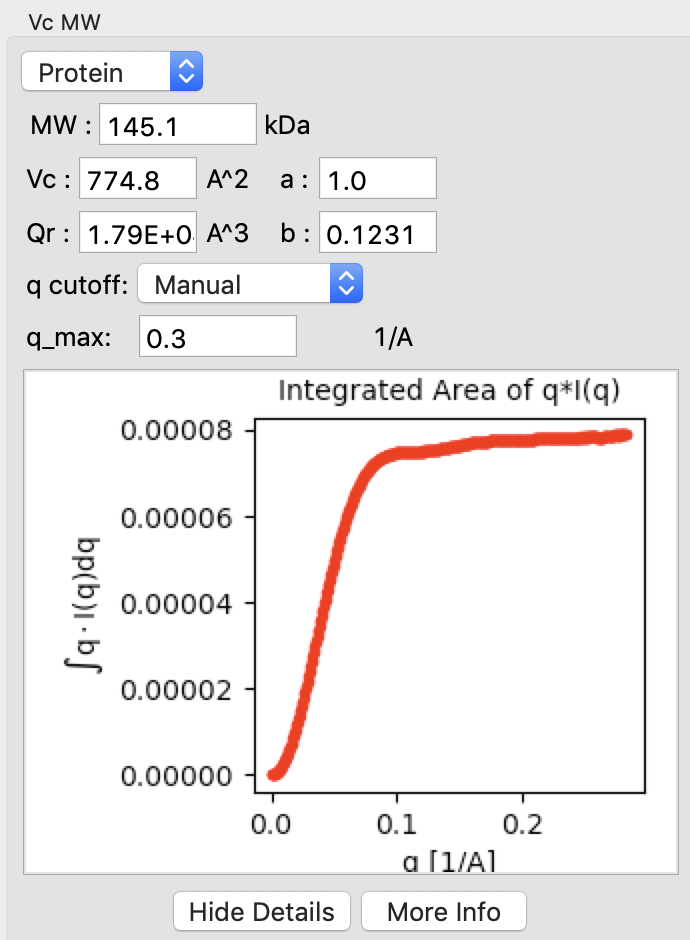
Click on the “Show Details” button for the Vc MW panel. You should see a graph, which shows the integrated area of \(qI(q)\) vs. q. For this method to be accurate, this value needs to converge at high q.
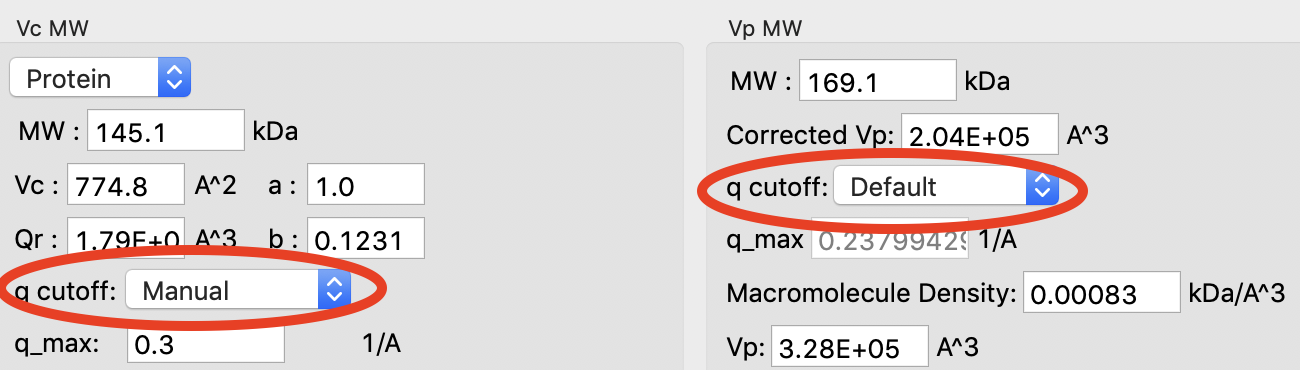
Click on the “Show Details” for the Vp MW panel. You’ll notice that both Vp MW and Vc MW have a “cutoff” selection. At higher q you can start to get scattering from flexibility or intra-molecular features that may reduce the reliability of the MW estimate. RAW automatically cuts off the scattering profile used based on the size of the object. You can change the cutoff if you need to.
Click the “OK” button to save your analysis.
Note: The “Cancel” button discards the analysis.
Tip: After clicking “OK” you can now click on the GI profile in the Profiles control panel and see the MW you just found in the Info panel.
Repeat the MW analysis for the lysozyme sample, which had a concentration of 4.27 mg/ml. The expected MW of lysozyme is 14.3 kDa.
Question: Does the Vc method work for the lysozyme data?