3D reconstruction with bead models – DAMMIF/N and DAMAVER in RAW
Shape reconstruction in SAXS is typically done using bead models (also called dummy atom models, or DAMs). The most common program used to generate these shapes is DAMMIF (and, to a lesser degree, DAMMIN) from the ATSAS package. We will use RAW to run DAMMIF/N. Because the shape reconstruction is not unique, a number of distinct reconstructions are generated, and then a consensus shape is made from the average of these reconstructions. The program DAMAVER from the ATSAS package is the most commonly used program for building consensus shapes. Note that you need ATSAS installed to do this part of the tutorial. Also, this tutorial uses ATSAS 3.1.1, some pieces may be slightly different on older versions of ATSAS, please see previous versions of this tutorial in that case.
This is not a tutorial on basic principles and best practices for doing bead model reconstructions. For that, please see the SAXS tutorial.
If you use RAW to run DAMMIF or associated programs, in addition to citing the RAW paper, please cite the papers given in the:
as appropriate.
A video version of this tutorial is available:
The written version of the tutorial follows.
Clear all of the data in RAW. Load the glucose_isomerase.out file that you saved in the reconstruction_data folder in a previous part of the tutorial.
Note: If you haven’t done the previous part of the tutorial, or forgot to save the results, you can find the glucose_isomerase.out file in the reconstruction_data/gi_complete folder.
Right click on the glucose_isomerase.out item in the IFT list. Select the “Bead Model (DAMMIF/N)” option.
Running DAMMIF generates a lot of files. Click the “Select” button for the output directory, make a new folder in the reconstruction_data directory called gi_dammif and select that folder.
Change the number of reconstructions to 5.
Note: It is generally recommended that you do 15-20 reconstructions. However, for the purposes of this tutorial, 5 are enough.
Note: For final reconstructions for a paper, DAMMIF should be run in Slow mode. For this tutorial, or for obtaining an initial quick look at results, Fast mode is fine.
Uncheck the “Refine average with dammin” checkbox.
Note: For final reconstructions for a paper, DAMMIN refinement should be done. However, it is quite slow, so for the purposes of this tutorial we won’t do it.
RAW can align the DAMMIF/N output with a PDB/mmCIF structure using CIFSUP from the ATSAS package. To do so, check the ‘Align output to PDB/mmCIF’ box and select the 1XIB_4mer.pdb file in the reconstruction_data/gi_complete folder.
Tip: If you’re not sure if you selected the correct file, hovering your mouse over the filename will show the full path to the file.
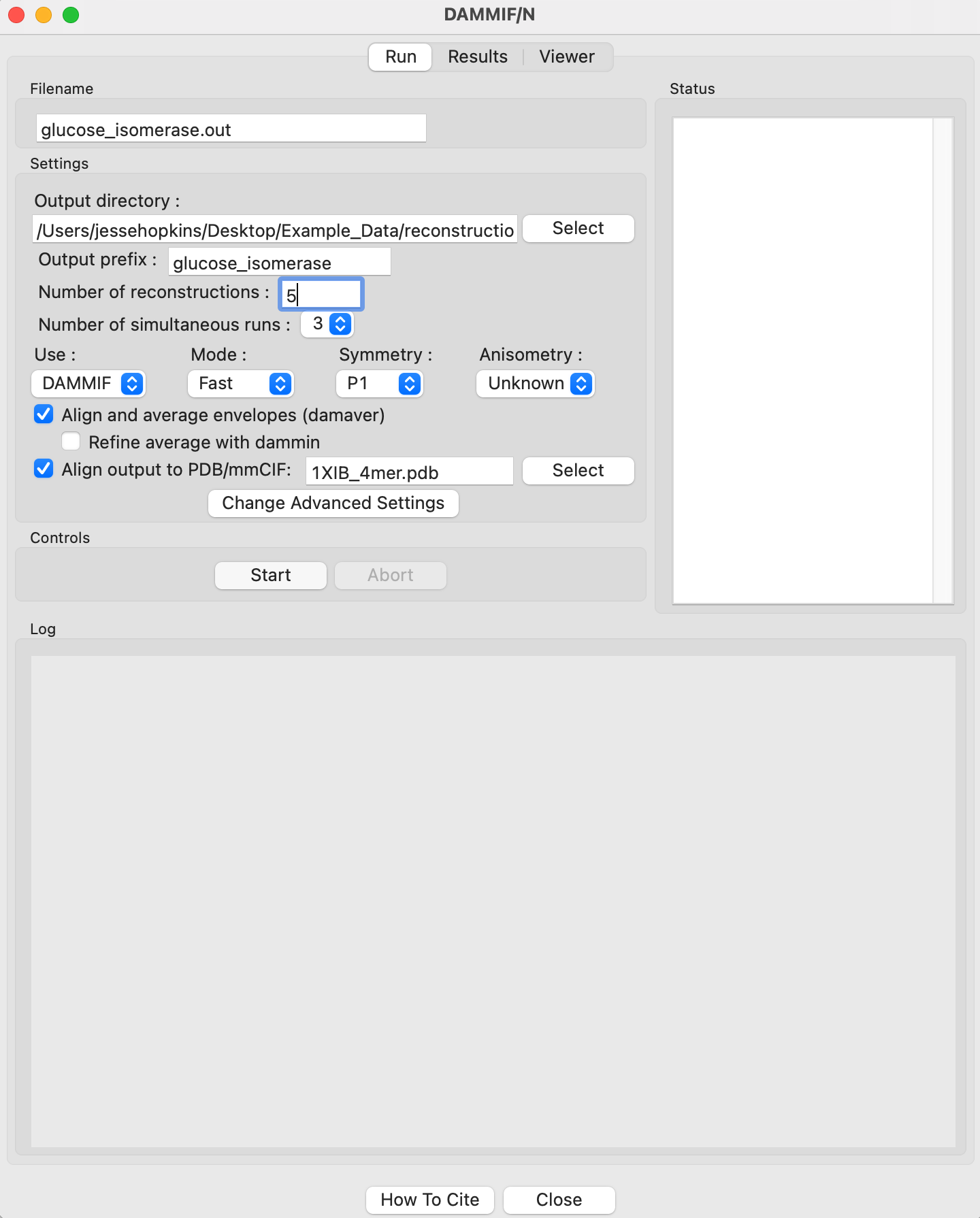
Click the “Start” button.
Note: The status panel will show you the overall status of the reconstructions. You can look at the detailed status of each run by clicking the appropriate tab in the log panel.
Note that by default the envelopes are aligned, clustered, and averaged using DAMAVER, and then the aligned and averaged profile is refined using DAMMIN.
Some settings are accessible in the panel, and all settings can be changed in the advanced settings panel.
Wait for all of the DAMMIF runs, DAMAVER, and alignment to finish. Depending on the speed of your computer this could take a bit.
Once the reconstructions are finished, the window should automatically switch to the results tab. If it doesn’t, click on the results tab.
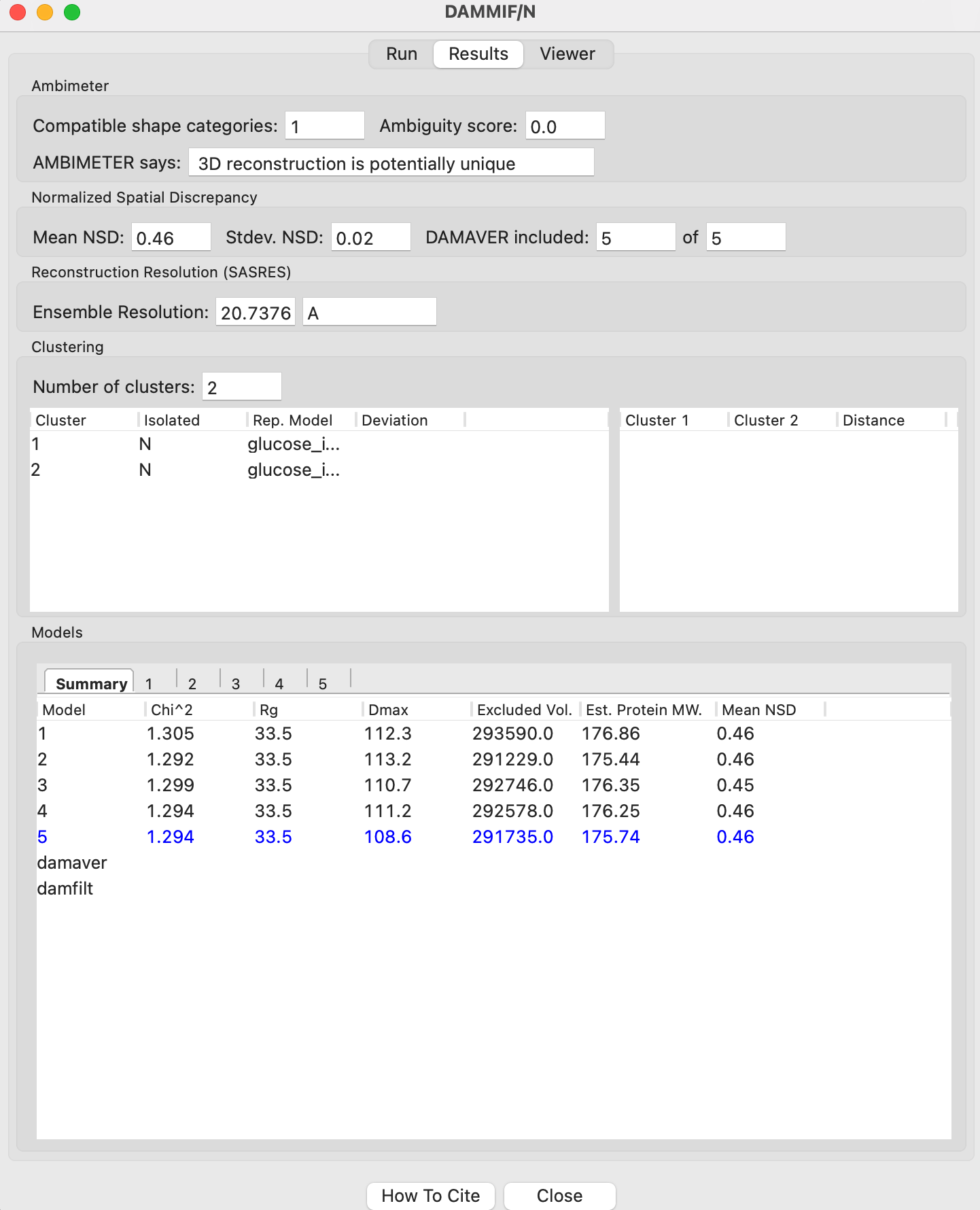
The results panel summarizes the results of the reconstruction run. At the top of the panel there is the AMBIMETER evaluation of how ambiguous the reconstructions might be (see previous tutorial section). If DAMAVER was run, there are results from the normalized spatial discrepancy (NSD), showing the mean and standard deviation of the NSD, as well as how many of the reconstructions were included in the average. If DAMAVER was run on 3 or more reconstructions, and ATSAS >=2.8.0 is installed, there will be the output of SASRES which provides information on the resolution of the reconstruction. If DAMAVER found more than one cluster, the number of clusters and information on each cluster is shown. Note that DAMCLUST (ATSAS <=3.1.0) provided more information about the clusters, so some fields will be blank with ATSAS >=3.1.1.
Information on each individual model is shown at the bottom. The summary tab gives the model chi squared, Rg, Dmax, excluded volume, molecular weight estimated from the excluded volume, and, if appropriate, mean NSD of the model.
Any models rejected from the average by DAMAVER will be shown in red in the summary tab list.
Tip: The model highlighted in blue in the summary tab is the ‘most probable’ model, this can be used as your final bead model instead of doing a DAMMIN refinement.
Note: DAMMIN in ATSAS >=3.1.0 doesn’t provide the Dmax value for the model.
Also, each individual model has a tab which shows the data, the model fit, and the residuals.
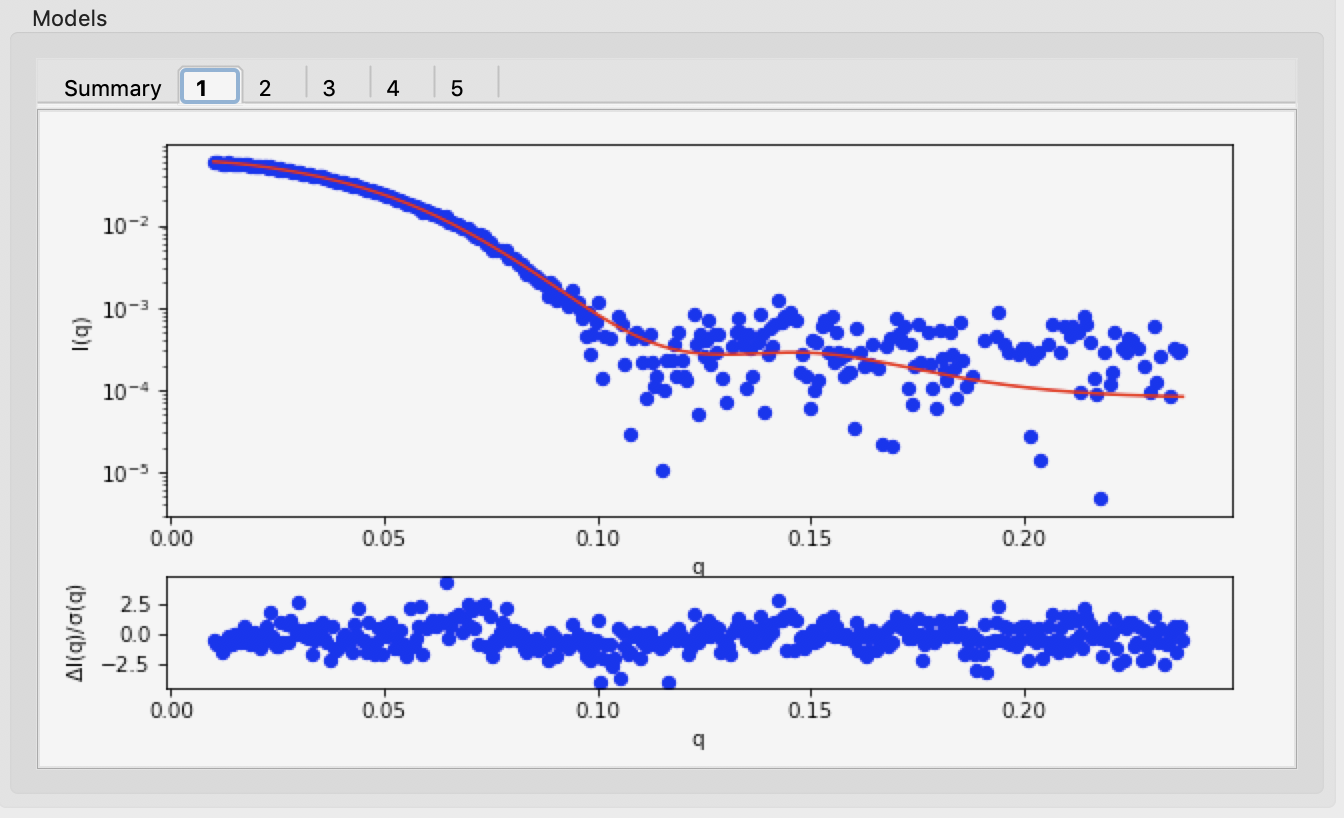
The results summary shown in Summary tab is automatically saved as a <prefix>_dammif_results.csv csv file, e.g. for this data as glucose_isomerase_dammif_results.csv. All the plots shown on the individual model tabs are automatically saved as a multi-page pdf file with the same name.
Click on the Viewer tab to open the model viewer.
Note: The model viewer is intended for a fast first look at the results. It is not currently up to the standards of a program like pyMOL.
Click and drag the model to spin it.
Note: For glucose isomerase, it should look more or less like a flattened sphere.
Right click and drag the model to zoom in and out.
Use the “Model to display” menu in the Viewer Controls box to change which reconstruction is displayed.
Click the “Close” button when you are finished looking at the results and reconstructions.
The results from individual DAMMIF runs are saved in the selected output folder with the name <prefix>_xx, where xx is the run number: 01, 02, etc. For this tutorial, that would be glucose_isomerase_01, glucose_isomerase_02, and so on. The different files produced are described in the DAMMIF manual.
Note: Generally, the file of interest is the -1.cif file, in this case glucose_isomerase_01-1.cif, glucose_isomerase_02-1.cif, etc.
If averaging was done with DAMAVER, the results are saved in the selected output folder with the given prefix, in this case glucose_isomerase. The output files generated are described in the DAMAVER manual.
Note: Generally, the file of interest is the generated damfilt mmCIF: <prefix>_damfilt.cif. For this tutorial, that would be glucose_isomerase_damfilt.cif.
If multiple clusters were found, the results are saved in the selected output folder with the given prefix (for this tutorial, glucose_isomerase). The files generated are described in the DAMAVER manual.
If refinement was done with DAMMIN, the results are saved in the selected output folder as refine_<prefix>, e.g. for this tutorial refine_glucose_isomerase. The files generated are described in the DAMMIN manual.
Note: Generally, the file of interest is the -1.cif file, in this case refine_glucose_isomerase-1.cif.
If alignment to a reference PDB/mmCIF was done with CIFSUP, the files aligned depend on what other processing was done.
If refinement was done, then there will be a single file named refine_<prefix>_-1_aligned.cif. For this tutorial, refine_glucose_isomerase-1_aligned.cif.
If no refinement is done but averaging is done, then the damaver and damfilt results are aligned, as well as the most probable model (the blue highlighted model in the summary panel). The associated filenames would be <prefix>_damaver_aligned.cif, <prefix>_damfilt_aligned.cif, and <prefix>_##_-1_aligned.cif where ## is the model number of the most probable model. For this tutorial, glucose_isomerase_damaver_aligned.cif, glucose_isomerase_damfilt_aligned.cif, and glucose_isomerase_##-1_aligned.cif.
If no refinement or averaging is done, then every calculated model is aligned. The associated filenames would be <prefix>_##-1_aligned.cif where ## is the model number of a model. For this tutorial, that is glucose_isomerase_##-1_aligned.cif.
You can save a summary table of the DAMMIF results with the pdf report that RAW can make. Close the DAMMIF/N window, right click on the glucose_isomerase.out item in the IFT control panel and select “Save report”. Use the “Add DAMMIF/N results .csv” button to add the glucose_isomerase_dammif_results.csv to the report. Then click “Save Report” and save the pdf report. If you open the report you will see a summary of the run parameters and numerical results saved as a table.
